 |
|||||||||||||||||||||||||||||||||||||||||||||||||||
What does it take to sequence the mammoth genome? Paleogenomics has become a reality only with the onset of next-generation sequencing. This technique overcomes problems that bedeviled Sanger sequencing, such as the necessity of cloning ancient DNA into vectors (DNA libraries), thus requiring a cloning host like E. coli to propagate it. Both steps are likely to introduce severe biases into the mixtures of ancient DNA to be analyzed. Next-generation sequencing, as marketed by Roche/454 Life Sciences, combines the two principles of emulsion PCR and pyro-sequencing to allow for a several-orders-higher throughput than what is possible on a Sanger capillary-electrophoresis DNA sequencer, while avoiding sequencing biases. It thus becomes feasible to sequence DNA from samples of fossil specimens that might contain only a small percentage of endogenous DNA. Below we provide a short tour of the technologies and instruments that are used in the mammoth genome project. |
|||||||||||||||||||||||||||||||||||||||||||||||||||
Principles of Roche 454 next-generation
sequencing
| |
In the following, the steps of the 454 bead-based sequencing technique are described,
which have been automated using a DNA sequencing instrument (Roche GS FLX)
and two reagent kits (LR).
|
|
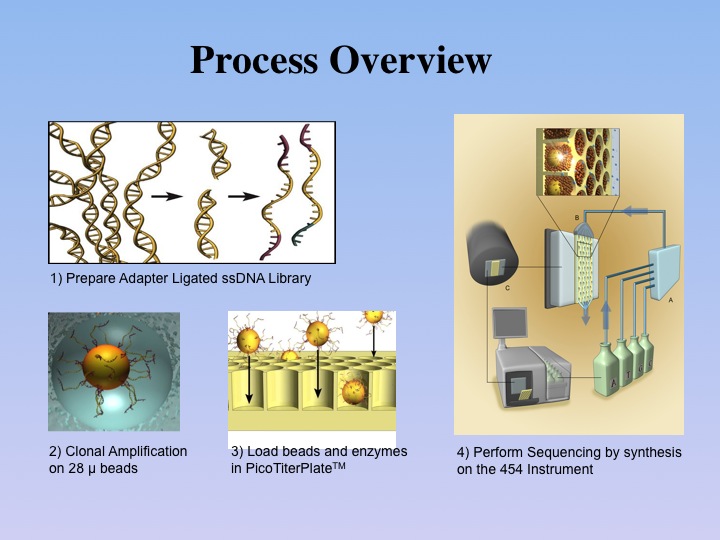 |
Once genomic DNA from a sample has been sufficiently
purified by our laboratory protocols, the double-stranded DNA is sheared
by various techniques to about 400-600 base pairs. After a quality-control
step, the DNA is converted into single strands to which short adaptors
are then ligated. The molecules are subsequently diluted to low concentration
and submitted to an emPCR reaction (emulsion-based PCR) together with
sepharose beads of 28 µm diameter. |
|
This process involves attaching
the single-stranded template DNA to the beads via the short adaptors, in
a hybridization reaction. Ideally, depending on the DNA and bead concentration,
a single bead is combined with a single DNA molecule. To allow for exclusive
amplification of just one DNA molecule, all beads are subsequently encapsulated
by lipid vesicles, because of the bi-phasic nature of the emulsion PCR. As
all required reagents are present in the emPCR solution, a standard PCR
amplification is now carried out on all beads in parallel in a single
solution, which amplifies the single DNA molecules attached to each of
the beads. After completion of the PCR reaction, the emulsion reactors
are broken and the beads are collected, each one now containing 107 identical
DNA molecules.
| |
1.6 million well PicoTiterPlate(TM),
which allows for the parallel sequencing of DNA segments separated in
different wells. |
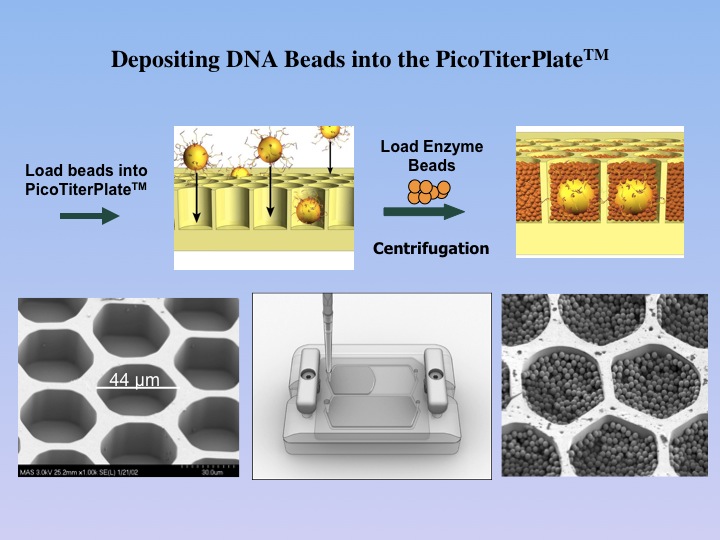 |
The
DNA-containing beads are then enriched and loaded
onto the PicoTiterPlate(TM) for a massively parallel sequencing analysis
of the attached DNA fragments. The sequencing reaction is based on the
detection of pyrophosphate, which is formed in the coupling process of
a nucleotide to an existing primer DNA by the DNA-polymerase. In
pyro-sequencing, the enzyme luciferase is used to catalyze
the hydrolysis of one pyrophosphate molecule into two ortho-phosphates
and one photon. The actual sequencing procedure is carried out in a series
of steps, in which the 4 nucleotides are individually cycled through a
flow chamber that consists of the bead-loaded PicoTiterPlate(TM) and the
optics of a CCD camera. Each nucleotide step is separated by a washing
step. This sequencing approach has also been termed sequencing by synthesis,
as each addition of a nucleotide to an existing oligonucleotide chain
is monitored by light emission. The recording software registers the amount
of light emitted in each reaction cycle, thus allowing for the assessment
of the number of nucleotides added per cycle. Since the sequencing reactions
are analyzed in a massively parallel fashion, up to 400,000 successful
reactions are recorded from a 70x70 mm PicoTiterPlate(TM), resulting in
20 to 40 Mb (million bases) of high quality data. |
|
Instead of the electropherograms
used for the display of DNA sequences created in Sanger-type sequencing,
the alternating reaction cycles of the 454 technology are recorded as
flowgrams (figure below). These allow assessment of how many nucleotides per
cycle were added to the growing DNA strand. |
Flowgram. In contrast to gel-based
sequencing, the 454 sequencing is based on detection of photons emitted
from individual wells in the 1.6 million well PicoTiterPlate(TM), as different
nucleotides are washed over the plate. Bases are identified when a photon is emitted
during polymerization using the a particular nucleotide;
multiple photons indicate homopolymer stretches of DNA. |
 As with Sanger sequencing,
each base determination is accompanied by a quality value. This allows
for assembly of the data using software provided by 454 Life
Sciences, which has been demonstrated to successfully assemble genomes
up to several Mb. |
|
Before the DNA can be sequenced
it needs to be analyzed in instruments that determine the purity and size of
the DNA molecules and the total amount available. These tests can be done by
either spectroscopy or electrophoresis. |
|
The Agilent Bioanalyzer assesses
the size and quantity of a DNA sample. |
 The DNA prepared for sequencing
has to undergo several hundred handling steps before it can be loaded
onto the DNA sequencer. |
 Skilled hands manipulate fractions
of small droplets containing DNA. |
 General molecular biology
laboratory for the DNA isolation and processing. |
 Two bead stations for the setup
of emPCR reactions. This is where a single DNA molecule is combined with
one |
28 µm bead inside a lipid vesicle.  The emPCR reactions need to
be thermo-cycled in PCR machines that vary the sample temperature according
to a computer program. |
 Roche GS FLX DNA sequencers
that were used for the project. |
 |
|
|
Computing the mammoth
genome. |
Analyzing the more than 4 billion base
pairs of the mammoth genome constitutes a huge computational problem, as
millions of DNA sequence comparisons need to be performed. The bioinformatics
part of the project therefore is as challenging as the generation of the
data itself. The team has used in-house software for most of these comparisons,
which will become part of ongoing and future projects for the analysis
of complete mammalian genomes. |
The Center for Comparative
Genomics and Bioinformatics operates several compute clusters for the
same general purpose: analyzing DNA sequence information. |
 The next-generation sequence
data is analyzed on clusters of Apple and Dell computers running the
Unix and Linux operating systems. |
 Dedicated compute clusters
are used for the processing of image data coming from the DNA sequencing
instruments. The results of these computations are large text files that
contain the four base pairs (in text format) that make up the genetic
information of any living being. |
 |
 |