 |
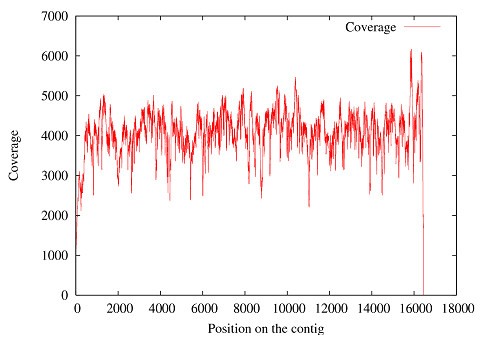
How accurate is the sequence data? We sequenced 4.12 Gb (billion bases) of data comprised of 32,876,587 reads with an average length of 125 bp. This data included high coverage (averaging 4430X) of the complete mitochondrial sequence, as shown in Figure 1, in addition to the DNA from the nuclear genome. |
 |
Figure
1: Read depth across the mitochondrial sequence of mammoth M4 |
We had determined the mitochondrial sequence in an earlier project, but having this extreme coverage had the benefit of allowing us to assess the sequencing error and the DNA damage rates for our sample by comparing individual reads with the consensus sequence. Following the method of Stiller et al. (2006) we calculated the DNA damage rate in the following manner: If we assume that the rate of T-to-C transitions is representative of the PCR enzymatic and sequencing error, then the true C-to-T DNA damage can be estimated by subtracting the number of T-to-C transitions from the number of C-to-T transitions. The same assumptions can be applied to A-to-G transitions, giving us the rate of true G-to-A DNA damage. Together these two (C-to-T and G-to-A) constitute the type 2 transitions, which have been shown to be the bulk of the DNA damage in the permafrost samples (Gilbert et. al. 2007). The differences between the consensus mtDNA and the trimmed reads covering it were calculated using a custom program and are categorized in Table 1. |
Table
1: The differences between mtDNA and trimmed reads for M4 (the mammoth sample that
provided most of our sequences). The consensus
base labels the rows and the sequencer-read base labels the columns. |
 |
| We identified 72,951,869 matches, 103,181 mismatches, 51,587 more bases in the consensus mtDNA (i.e. cases where the read erroneously left out a base, almost always in a homopolymer run), 97,661 more bases in the reads (again mostly in homopolymer regions). These results were then used to calculate the various damage rates, and the results of the analysis are listed in Table 2. |
Table
2: |
 |
References: Stiller, M. et al. (2006) Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA. Proc. Natl. Acad. Sci. USA 103:13570-13584. Gilbert, M.T.P., J. Binladen, W. Miller, C. Wiuf, E. Willerslev, H.Poinar, J. Carlson, J. Leebens-Mack, and S.C.Schuster (2007) Recharacterization of ancient DNA miscoding lesions: Insights in the era of sequencing-by-synthesis. Nucleic Acids Res. 35:1-10. |
 |